がんというエコシステムにおける
抗腫瘍免疫応答
がん細胞は正常な遺伝子に変異が生じ、その変異が修復されないことで発生します。こうした遺伝子には必要な時に細胞を増殖させる遺伝子(がん遺伝子)、不必要な時には増殖を止める遺伝子(がん抑制遺伝子)があり、これらの変異によって細胞が異常に増殖し、血管などを通して新たな場所に移動し、さらに増殖します。生体には本来細胞ががん化した際に、それらを排除するための免疫監視のメカニズムが備わっていますが、その仕組みの破綻ががんの発症・進展に大きく寄与しています。近年、がん細胞を認識し直接攻撃して殺す能力を持つT細胞に関する研究が著しく進展しており、免疫チェックポイントと呼ばれるPD-1やPD-L1を標的にし、それらの活性化を抑制することによって、T細胞ががん細胞を攻撃する能力を維持・回復する免疫療法が様々な種のがんにおいて非常に有効であることが示されています[1]。しかし一方でおよそ半数のがん患者さんでこうした免疫療法が効かないという事実も存在し、過剰な自己免疫応答による大腸炎、腎機能障害などの副作用(immune-related Adverse Events, irAE)等も見られることから[1]、がん細胞に対する免疫応答の基礎的な理解が、がん免疫療法のさらなる発展に重要と考えられます。
遺伝情報をコードするDNAががん細胞に対する免疫応答を惹起する
T細胞を活性するためには免疫チェックポイント分子の阻害のみならず様々な免疫応答が重要です。中でもマクロファージや樹状細胞などの免疫細胞ががん細胞由来の抗原を認識してT細胞に抗原提示するとともに、T細胞を活性化するサイトカインを産生する仕組み(自然免疫応答)の制御が、がん免疫療法の新たな戦略として注目されています。DNAやRNAといった核酸は遺伝情報の伝達物質ですが、細菌やDNA/RNAウイルス由来の核酸は哺乳類の核酸とは異なる特有の配列や修飾を持っており、これらを特異的に認識するシグナル伝達経路は古くから研究されてきました[2]。一方でそうした非自己由来の核酸のみならず、自己の細胞由来の核酸も抗原として働き自然免疫細胞を活性化することが明らかになり、こうした免疫応答の制御異常ががんや自己免疫疾患など様々な病気の原因となることがわかってきました。
cGAS-STING経路は細胞内のDNAを認識する主要な経路で、免疫細胞のみならず、線維芽細胞やがん細胞といった様々な細胞に発現し、I型インターフェロン(IFN-I)などの抗腫瘍免疫応答を増強するサイトカインの産生を誘導します[3]。従来DNAは核内に存在していますが、DNA損傷によって生じた自己のDNAや病原体由来のDNAが細胞質内に取り込まれると、DNAセンサーであるcGASがこれらを認識し、セカンドメッセンジャーであるcGAMPを産生します。下流のアダプター分子であるSTINGがこのcGAMPを認識して活性化すると、小胞体からゴルジ体へ移行してリン酸化酵素であるTBK1と結合し、転写因子IRF3を核内に移行させ、I型インターフェロンの遺伝子発現を誘導します(図1)。

I型インターフェロンは微小環境に存在する様々な細胞に働き、がん細胞に対する攻撃を活性化します[4]。例えば抗原提示細胞である樹状細胞に対してはその成熟を促進するとともに、がん抗原特異的T細胞を活性化して、がん微小環境に誘導します。またこれらのT細胞やNK細胞に直接働いてがん細胞に対する殺傷能力を高めます。一方でがんに対する攻撃を抑え込む制御性T細胞に対してはその免疫抑制能力を抑制する働きもあります。近年新たながん免疫療法の戦略として、がん微小環境でI型インターフェロンの産生を誘導する目的で、cGAS-STING経路の主要分子であるSTINGのアゴニストが複数の製薬会社で開発されており、その効果を調べるため多くの臨床治験が進行中です[5]。
がんというエコシステムにおけるcGAS-STING経路の活性化
これまでcGAS-STING経路は主に樹状細胞やマクロファージといった自然免疫細胞で活性化されることが知られていました。これらの免疫細胞は死んだがん細胞を貪食し、そのDNAを取り込むことによってcGAS-STING経路を活性化し、I型インターフェロンを産生します[6]。一方でがん微小環境に存在する免疫細胞以外の細胞でもcGAS-STING経路が働き、抗腫瘍免疫応答に寄与することがわかってきました(図2)。
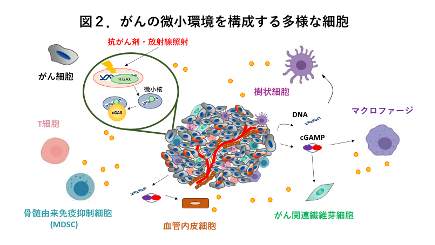

興味深いことに、がん細胞においてゲノムの不安定性による細胞分裂の異常や、抗がん剤や放射線照射で引き起こされるDNAダメージによって、細胞質内に微小核と呼ばれる構造が出現し、DNAセンサーであるcGASを活性化することが報告されました[7, 8]。さらに、がん細胞自身がエクソソームと呼ばれる小胞などを介して、STINGの内在性アゴニストであるcGAMPを細胞外に放出し、隣接する細胞間で形成されるギャップ結合や細胞膜に存在するトランスポーターといったシステムによってcGAMPを輸送することで、がん微小環境にいる細胞を活性化することが示されています(図3)。この受け取り側の細胞は自然免疫細胞に限らず、血管内皮細胞[9]やがん関連線維芽細胞[10]といった、従来免疫応答を担うとは考えられていなかった様々な細胞が関与していることが分かりました。すなわちcGAS-STING経路は古典的な自然免疫細胞のみならずがん微小環境を構成する様々な細胞で活性化され、がんというエコシステムにおいて抗腫瘍免疫応答に重要な役割を果たしているのです(図3)。また抗がん剤や放射線治療は従来がん細胞のDNAを破壊し、がん細胞を殺す目的で利用されてきましたが、実は免疫応答を活性化することによってもがんの縮小に貢献しているという新たな側面が見えてきました。
抗腫瘍免疫応答に対するがんの攻防・がん細胞の進化と免疫抑制
一方がん細胞は自身の生存・増殖を妨げるこれらの免疫応答を回避するため、様々な戦略を駆使しています。前述しましたように、がん細胞の特徴として染色体やゲノムの不安定性、それに付随する遺伝子異常があげられます。黒色腫や肺がん、大腸がんなどヒトの多くのがんではcGASやSTINGの発現が低下しており、それらの活性化を妨げる変異が起きています[11, 12]。この発現の低下はエピジェネティクス制御によるもので、例えば乳がんではがん遺伝子であるMYCがヒストンのメチル化を制御するDNMT1の発現を誘導し、STINGのプロモーター領域のメチル化を促進することで遺伝子発現を抑制していることが報告されています[13]。またcGAS-STING経路が保存されているがん細胞においても、ゲノムの不安定性で生じた微小核によるcGASの活性化が脳腫瘍や乳がんなど一部のがんの転移に寄与していることがわかりました[14, 15]。つまりがんの種類や原発巣・転移巣などの違いによって、あるシグナル伝達経路の活性化ががんの促進に働くのか抑制に働くのか異なるということになり、非常に興味深いところです。
またこれまではがん細胞の増殖という観点のみで注目されていたがん遺伝子およびがん抑制遺伝子に、実は免疫応答を制御する機能があることがわかってきました。例えば乳がんや胃がんの患者さんでよくみられるがん遺伝子HER2の増幅はがん細胞の増殖を促進させることが知られていますが、近年HER2はSTINGと直接結合してその活性化を抑制することが報告されました[16]。また、がん抑制遺伝子であるDAPK3は肺がんや卵巣がん、大腸がんでその酵素活性を消失させる変異が見つかっており[17]、がん細胞におけるこれらの変異はがん細胞の増殖を促進する作用を持つ一方、cGAS-STING経路の活性化を抑制することがわかりました[18]。一方で機能的なDAPK3はマクロファージなどの自然免疫細胞や血管内皮細胞でもcGAS-STING経路を制御していることから、がん細胞以外において正常な機能を持つがん抑制遺伝子ががんの増殖を抑制するという新たな側面が明らかになりました[18]。こうした制御はcGAS-STING経路に限ったものではなく、がん遺伝子MYCが免疫チェックポイント分子であるCD47やPD-L1の発現を増加させることで免疫細胞からの攻撃を免れる、がん遺伝子Krasの活性化やがん抑制遺伝子PTENの喪失により抗腫瘍免疫応答を抑制するサイトカインの産生が促進されるなど数多くの報告があります[19](図4)。すなわち、がん細胞は遺伝子変異によって異常な増殖能力を獲得するとともに、自身の免疫応答やがん微小環境を構成する細胞の免疫応答を抑制することによって免疫系による除去を回避しているのです。

おわりに
近年非常に発達しているシークエンス技術により、がん組織における多様性を一細胞あたりで解析できるようになり、患者さん一人一人、また同一の腫瘍組織内における詳細な細胞群の解析、遺伝子発現・遺伝子変異のプロファイルが可能になりました。上述したように、がん細胞は異なるがん遺伝子およびがん抑制遺伝子の変異を組み合わせて抗腫瘍免疫応答を回避しており、さらに微小環境から受ける選択圧によって免疫細胞による攻撃を回避できるがん細胞が選択される仕組みが明らかになってきました[20]。すなわちがんの進化の一端は免疫編集が担っており、これらを踏まえ、がんの種類・ステージおよび微小環境の構成の違いによりどのような分子標的薬が効くのかという効果予測や、個々の免疫療法に対する反応性や耐性の機序の解明を発展させることが今後の課題であると考えられます。
注:図の一部はBioRender.comを用いて作成しました。
1. Bagchi, S., R. Yuan, and E.G. Engleman, Immune Checkpoint Inhibitors for the Treatment of Cancer: Clinical Impact and Mechanisms of Response and Resistance. Annu Rev Pathol, 2021. 16: p. 223-249.
2. Schlee, M. and G. Hartmann, Discriminating self from non-self in nucleic acid sensing. Nature Reviews Immunology, 2016. 16(9): p. 566-580.
3. Chen, Q., L. Sun, and Z.J. Chen, Regulation and function of the cGAS-STING pathway of cytosolic DNA sensing. Nat Immunol, 2016. 17(10): p. 1142-9.
4. Zitvogel, L., et al., Type I interferons in anticancer immunity. Nat Rev Immunol, 2015. 15(7): p. 405-14.
5. Kong, X., et al., STING as an emerging therapeutic target for drug discovery: Perspectives from the global patent landscape. Journal of Advanced Research, 2022.
6. Corrales, L., et al., The host STING pathway at the interface of cancer and immunity. J Clin Invest, 2016. 126(7): p. 2404-11.
7. Harding, S.M., et al., Mitotic progression following DNA damage enables pattern recognition within micronuclei. Nature, 2017. 548(7668): p. 466-470.
8. Mackenzie, K.J., et al., cGAS surveillance of micronuclei links genome instability to innate immunity. Nature, 2017. 548(7668): p. 461-465.
9. Demaria, O., et al., STING activation of tumor endothelial cells initiates spontaneous and therapeutic antitumor immunity. Proc Natl Acad Sci U S A, 2015. 112(50): p. 15408-13.
10. Arwert, E.N., et al., STING and IRF3 in stromal fibroblasts enable sensing of genomic stress in cancer cells to undermine oncolytic viral therapy. Nat Cell Biol, 2020.
11. Xia, T., H. Konno, and G.N. Barber, Recurrent Loss of STING Signaling in Melanoma Correlates with Susceptibility to Viral Oncolysis. Cancer Res, 2016. 76(22): p. 6747-6759.
12. Xia, T., et al., Deregulation of STING Signaling in Colorectal Carcinoma Constrains DNA Damage Responses and Correlates With Tumorigenesis. Cell Rep, 2016. 14(2): p. 282-97.
13. Wu, S.-Y., et al., MYC suppresses STING-dependent innate immunity by transcriptionally upregulating DNMT1 in triple-negative breast cancer. Journal for ImmunoTherapy of Cancer, 2021. 9(7): p. e002528.
14. Bakhoum, S.F., et al., Chromosomal instability drives metastasis through a cytosolic DNA response. Nature, 2018.
15. Chen, Q., et al., Carcinoma–astrocyte gap junctions promote brain metastasis by cGAMP transfer. Nature, 2016. 533(7604): p. 493-498.
16. Wu, S., et al., HER2 recruits AKT1 to disrupt STING signalling and suppress antiviral defence and antitumour immunity. Nature Cell Biology, 2019. 21(8): p. 1027-1040.
17. Brognard, J., et al., Cancer-associated loss-of-function mutations implicate DAPK3 as a tumor-suppressing kinase. Cancer Res, 2011. 71(8): p. 3152-61.
18. Takahashi, M., et al., The tumor suppressor kinase DAPK3 drives tumor-intrinsic immunity through the STING–IFN-β pathway. Nature Immunology, 2021. 22(4): p. 485-496.
19. Wellenstein, M.D. and K.E. de Visser, Cancer-Cell-Intrinsic Mechanisms Shaping the Tumor Immune Landscape. Immunity, 2018. 48(3): p. 399-416.
20. Rosenthal, R., et al., Neoantigen-directed immune escape in lung cancer evolution. Nature, 2019. 567(7749): p. 479-485.
執筆者:高橋まり子
2013年 東京大学医科学研究所にて博士号取得。2014年から2020年までLa Jolla Institute for ImmunologyにてPostdoctoral Fellow。2021年からMassachusetts General Hospital/Harvard Medical SchoolにてResearch Staff (現職)。ケミカルバイオロジーを用いて抗腫瘍免疫応答を異なる視点から理解し、新しいがん免疫療法につなげる道筋を模索中。